2020A13 Describe
the time course between an intravenous injection of a general
anaesthetic agent to loss of consciousness. Explain the delay using
pharmacokinetic principles.
List:
· Intro
· Summary
· Kinetics/dynamics
· Dynamics
· Special populations
Intro:
|
IV induction |
· Agent induces unconsciousness in one arm-brain circulation time · Unconsciousness occurs upon reaching a threshold concentration at the effect site, usually well before the peak |
|
Problems |
· Three compartment model poorly describes induction kinetics · Variable relationship between dose and concentration · Variable relationship between concentration and effect |
|
Implications |
· Risk of overdose -> hypotension · Risk of underdose -> risk of awareness · Propofol TCI models are inaccurate at induction |
Summary:
|
Kinetics |
· Delay = administration -> plasma concentration (Cp) · Not well represented by the three compartment model · Mostly patient-dependent delay |
|
Biophasics |
· Delay = plasma concentration -> effect site concentration (Ce) · Represented by t1/2ke0 · Inferred by lag between ∆Cp and ∆EEG pattern · Mostly drug-dependent delay |
|
Dynamics |
· Delay = effect site concentration -> effect · Represented by dose response curve · Effects on ionotropic receptors brainstem, thalamus, cerebral cortex · Effects are virtually immediate · Mostly threshold-dependent delay |
Kinetic/biophasic determinants:
|
Graph |
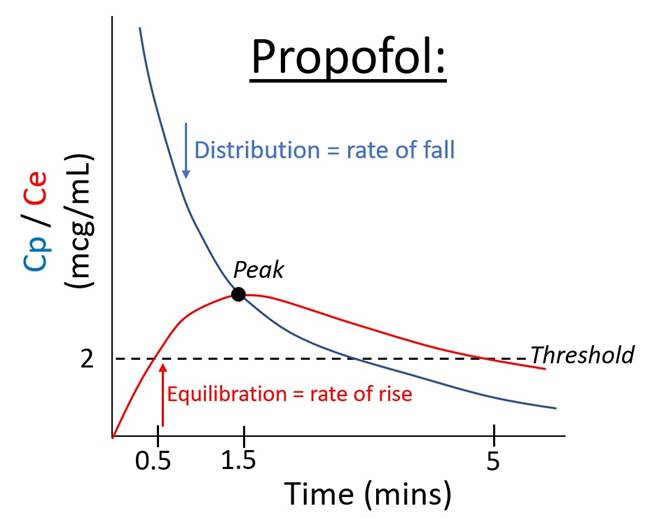
Speed of LOC ∝ (magnitude of peak effect) / (time to peak effect) |
|
Magnitude of peak |
Peak∝ · ↑Dose size · ↑Speed of injection (bolus cf. TCI injection) · ↓Cardiac output (↑pregnant/neonate/obese, ↓elderly/shock) · ↓Central blood volume (↑pregnant/obese/neonate, ↓elderly/shock) · ↑Recirculatory second peak (important if bolus is slow) |
|
Time to peak |
Speed ∝ · ↑Rate of delivery to effect site o ↑Cardiac output (note bivalent) o ↓Distance from injection site (e.g. jugular cf. foot) · ↑Rate of distribution o ↑Cardiac output (note bivalent) o ↑Compartment uptake (e.g. ↑lipid solubility) · ↑Rate of effect site equilibration
o ↑Lipid solubility (e.g. thiopentone: t1/2ke0 1 min -> clear endpoint) o ↑% Unionized (e.g. propofol >99%: t1/2ke0 2.6 mins) o ↓Thickness (e.g. immature BBB in neonate) |

Pharmacodynamic determinants:
|
Physiological |
· Neonate: immature brain structures and pathways -> ↓Cp50 · Elderly: ?↓ion channel function, ?↓ synaptic activity · Pregnancy: progesterone -> ↓Cp50 · Obesity: inflammatory cytokines -> ↓Cp50 |
|
Pathological |
↓Cp50 if: · ↓mAP (<40mmHg) · ↓pO2 (<40mmHg) · ↑pCO2 (>60mmHg sedation, >80mmHg anaesthesia if acute) · ↓Temp · ↓pH ↑Cp50 if: · Anxiety, ↑SNS · ↑Temp |
|
Drug interactions |
· Synergistic: e.g. fentanyl 1mcg/kg reduces dose of propofol by 20% · Additive: e.g. ↓propofol Cp50 if co-induction with volatile agent · Infra-additive: e.g. ketamine + midazolam · Antagonistic: e.g. propofol + acute amphetamines · Tolerance: chronic barbiturate use -> ↑Cp50 |
|
Pharmacogenomics |
· e.g. propofol Cp50 for immobility is 15mcg/mL with std dev 5mcg/mL · Polymorphism of receptors, ion channels, ICF signalling |
Special populations:
|
|
Changes |
Time to peak |
Magnitude of peak |
|
Neonate |
↑CO +100% ↑%CO to brain ↓Arm-brain distance |
↓ |
↓ |
|
Pregnant |
↑CO +25% ↓%CO to brain ↑BV 50% |
↓ |
↓ |
|
Elderly |
↓CO variable ↓BV variable ↑%CO to brain variable |
↑ |
↑ |
|
Shocked |
↓CO ↓BV ↑%CO to brain |
↑ |
↑ |
Feedback welcome at ketaminenightmares@gmail.com